






Когда-то таких больных объявляли оборотнями, пособниками дьявола и сжигали на кострах. Или прятали в заточении. Сегодня их, конечно, никто не преследует, но сами болезни зачастую вынуждают людей к изоляции. Остаётся надеяться, что достижения генетики когда-нибудь помогут таким людям выздороветь.
Есть болезни распространённые, есть те, что встречаются крайне редко (не более 10 случаев на 100 тыс. человек). Но бывают и такие, с которыми далеко не каждому врачу доводилось столкнуться. Мы решили рассказать о самых удивительных из них.
Бородавочная эпидермодисплазия
Таких пациентов всего несколько десятков в мире. Болезнь превращает кожу в подобие древесной коры. Причина в аномальной восприимчивости организма к вирусу папилломы человека (ВПЧ). Чаще всего наросты покрывают конечности, но могут быть также на лице и теле. Пластическая операция лишь ненадолго убирает симптомы – спустя время они появляются вновь.
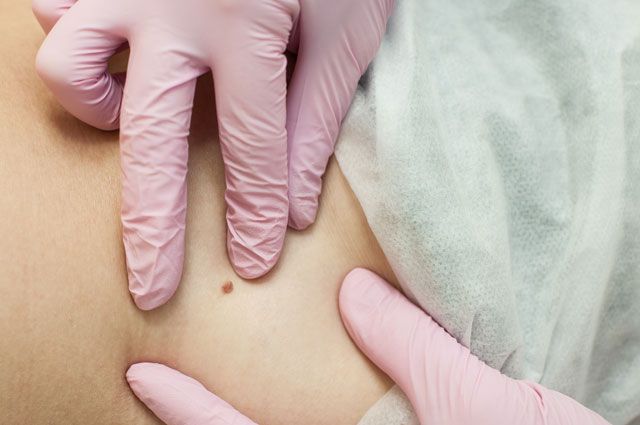
Пигментная ксеродерма
В народе эту болезнь прозвали «вампирской», ведь её главный симптом – непереносимость солнечных лучей. Кожа таких больных лишена естественной защиты от ультрафиолета. Недуг проявляется рано – в 2–3 года, а затем лишь усиливается. Раздражение, трещины, неравномерная пигментация, деформация лица и ушей, а со временем атрофия и рак кожи – вот что ждёт пациента, если он не будет избегать солнца и защищать кожу специальными средствами. Из-за светобоязни пациенты предпочитают вообще не видеть белого света и выходят на прогулки по ночам. Если полностью исключить контакт с УФ-излучением, можно прожить долго.
Ихтиоз
При этом генетически обусловленном обменном нарушении (болеет 1 человек из 30 тыс.) нарушается синтез кератина – белка, из которого состоит кожа. Эпидермис грубеет, утолщается, покрывается трещинами и чешуйками. Лицо деформируется: веки теряют возможность моргать, рот растягивается в улыбке. Дети с такой тяжёлой патологией обычно умирают в младенчестве, лишь каждый пятый может дотянуть до подросткового возраста. Немного облегчить страдания больным помогают многократные приёмы ванны и постоянное увлажнение кожи, а также лечебное питание и препараты, стимулирующие иммунитет.

Аквагенная крапивница (аллергия на воду)
Одну из самых редких форм аллергии (зарегистрировано менее сотни пациентов, чаще женщины) открыл дерматолог Уолтер Шелли в 1964 г. Она возникает при контакте с водой. Симптомы могут быть разные. Чаще это волдыри, похожие на ожог крапивой, кожная сыпь, но возможны головная боль, одышка, кашель и пр. К счастью, пить воду пациенты могут, поскольку слизистые оболочки на аллерген не реагируют, только кожа.
Фатальная семейная бессонница
А вот при этой редчайшей наследственной нейродегенеративной болезни человек не может уснуть, даже несмотря на самые сильные снотворные, в результате чего умирает через полгода (максимум через 2–3 года). Данный диагноз зафиксирован всего в 40 семьях по всему миру. Патологию открыл врач Игнацио Ройтер в 1979 г., наблюдая смерть от этой болезни родственника жены. Как правило, смертельная бессонница возникает в 30–60 лет. Её причина – поломка в кодирующем веществе 178-го гена PRNP, расположенного в 20-й хромосоме, где вместо аспарагина синтезируется аспарагиновая кислота. Из-за этого здоровый белок превращается в агрессивный прион, который нарушает проводимость нейронных импульсов и приводит к необратимым изменениям.

Анальгия
В редких случаях из-за мутации в гене SCN9A у людей может возникать врождённая нечувствительность к боли. Чаще всего патология развивается у детей до 2 лет и проходит с годами. Но за такими детьми нужен глаз да глаз, потому что, не зная боли, они ничего не опасаются, рискуя получить травмы и ожоги.
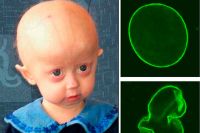
Прогерия (синдром Хатчинсона – Гилфирда)
Сетуете, что жизнь коротка? Тогда что же говорить о людях с прогерией, редкой генетической болезнью (всего около 350 зарегистрированных случаев), у которых голова лысеет, а лицо и тело покрываются морщинами уже в раннем возрасте. Эта болезнь поражает одного человека из 4–7 млн. Причина – спонтанная мутация в одной из двух копий гена LMNA. Болезнь диагностируется, как правило, к двум годам, когда у ребёнка наблюдаются задержка роста, ускоренное развитие верхней части черепа и другие симптомы. Такие пациенты живут в среднем 13 лет (максимум 25–27). К этому возрасту их организм уже изношен, как у столетних старцев, поэтому юные по паспорту люди умирают от инфарктов, инсультов, атеросклероза.
Синдром Юнера Тана
Эту болезнь открыл турецкий исследователь, обнаруживший в сельской местности семью Улас, все члены которой – пятеро братьев и сестёр – передвигались на четвереньках. Проблем с опорно-двигательным аппаратом у них не было, пальцы отлично работали (женщины в семье вязали крючком и вышивали). Тем не менее эти люди не могли стоять прямо, поэтому биолог решил, что имеет дело с откатом в эволюции человека. Но оказалось, во всём виновата поломка гена VLDLR, отвечающего за развитие мозжечка, который управляет координацией движений и равновесием.
Кстати
Редких (орфанных) заболеваний немало – около 7 тысяч. Тем не менее об этих патологиях мы мало что знаем. Разве что одной из них заболеет известный человек, как случилось с боковым амиотрофическим склерозом – болезнью Стивена Хокинга. Или если вдруг появится лекарство, как произошло со спинальной мышечной атрофией (СМА). В остальное время мы гоним мысли об этих патологиях, ведь большинство их труднодиагностируемы и, как правило, неизлечимы.

Комментарий эксперта
 Главный детский аллерголог-иммунолог, научный руководитель областного центра детской аллергологии и иммунологии Научно-исследовательского клинического института детства Московской области, доктор медицинских наук, профессор Андрей Продеус:
Главный детский аллерголог-иммунолог, научный руководитель областного центра детской аллергологии и иммунологии Научно-исследовательского клинического института детства Московской области, доктор медицинских наук, профессор Андрей Продеус:
– Расширение неонатального скрининга, которое с этого года вводится в стране, – идеологическая революция в медицине. Младенцев будут проверять более чем на 40 генетических заболеваний. Ведь, вместо того чтобы лечить тяжёлую генетическую патологию, можно просто не дать ей клинически проявить себя или сделать эти проявления минимальными.

До сих пор такой скрининг в роддомах проводился формально, ведь сегодня практикуется ранняя выписка. А на скрининг в поликлинику по месту жительства либо доходят не все, либо делают это с опозданием. Но в случае, например, со спинальной мышечной атрофией (СМА), когда моторные нейроны у больного ребёнка отмирают ежедневно, промедление даже на месяц может быть фатальным, поскольку эффективность запоздалого лечения сводится к нулю. Болезни обмена веществ, такие как фенилкетонурия, и вовсе не проявляют себя напрямую. Пациент медленно деградирует, потому что токсины отравляют его головной мозг. И когда диагноз ставится, может быть уже слишком поздно – деградация мозга необратима.
Теперь скрининг у доношенных детей будет делаться в течение 24–72 часов после рождения, у недоношенных – на 7-е сутки жизни.



